Drs Israa Al Shawi, FICMS, & Ali Al Hilaly, DVD
Al_Hashmia Hospital
Babylon,Iraq
Abstract: 14 to boy with photosensitivity and verrucous skin lesions.
HPI: This 14 yo boy's story started when he was two months old. He developed many bullae on scalp and extremities which healed spontaneously leaving a thickened skin. Over the years, he also developed many dark brown lesions in a generalized distribution. He has two sisters (age three and five) and two young cousins (male and female) with the same features. His parents are cousins and they are unaffected. There are a sister and brother who are normal
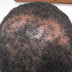
O/E: Large numbers of brown-black verrucous papules and plaques distributed all over the body. These lesions resemble seborrheic keratosis. He has erythema of sun-exposed areas and thick brown scales of scalp and sides of the face with alopecia. His body hair is coarse and is reported to sometimes improve spontaneously.
Clinical Photos:




Lab: None available at present
Pathology: One verrucous papule showed hyperkeratosis, acanthosis, papillomatosis and intranuclear inclusions consistent with verruca vulgaris.
Diagnosis: Not clear
Questions: This appears to be an autosomal recessive disorder.
If we consider it as Epidermodysplasia verruciformis, what is the explanation for scarring alopecia, photosensitivity and hypertrichosis?
Could this be a patient with Rothmund-Thompson Syndrome?
What are your opinions about the diagnosis?
What further studies can be done to establish the diagnosis, or can this be made clinically?
If genetic testing is indicated, what tests should be done and what kind of samples would they need?
References:
Rothmund-Thomson Syndrome [Internet].
Wang LL, Plon SE.
In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-.
1999 Oct 06 [updated 2009 Apr 07].
Excerpt
Disease characteristics. Rothmund-Thomson syndrome (RTS) is characterized by poikiloderma; sparse hair, eyelashes, and/or eyebrows/lashes; small stature; skeletal and dental abnormalities; cataracts; and an increased risk for cancer, especially osteosarcoma. The skin is typically normal at birth; the rash of RTS develops between age three and six months as erythema, swelling, and blistering on the face and subsequently spreads to the buttocks and extremities. The rash evolves over months to years into the chronic pattern of reticulated hypo- and hyperpigmentation, punctate atrophy, and telangiectases, collectively known as poikiloderma. Hyperkeratotic lesions occur in approximately one-third of individuals. Skeletal abnormalities include dysplasias, absent or malformed bones (such as absent radii), osteopenia, and delayed bone formation. Diagnosis/testing. The diagnosis of RTS is established by clinical findings — in particular, the characteristic rash. Routine cytogenetic studies of lymphocytes or skin fibroblasts may reveal mosaic abnormalities of chromosome 8, such as trisomy 8, partial 8q duplication, and tetrasomy 8q, which have been seen in individuals with RTS but are not diagnostic. Skin biopsy may show poikilodermatous changes, which are nonspecific but consistent with RTS. RECQL4 is the only gene associated with RTS to date; although evidence suggests genetic heterogeneity, no other locus for RTS has been identified. Molecular testing of RECQL4 is clinically available. Management. Treatment of manifestations: pulsed dye laser to treat the telangiectatic component of the rash; surgical removal of cataracts; and standard treatment for cancer. Prevention of secondary complications: use of sunscreens with both UVA and UVB protection to prevent skin cancer. Surveillance: annual physical and eye examination, monitoring of skin for lesions with unusual color or texture, screening for osteosarcoma. Agents/circumstances to avoid: excessive sun exposure. Genetic counseling. RTS is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once an at-risk sib is known to be unaffected, the risk of his/her being a carrier is 2/3. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible if the disease-causing mutations in the family are known.














0 comments:
Post a Comment